
Carbocation Stability Order: The Definitive Guide
A carbocation is an sp² trigonal planar species with only 6 valence electrons and an empty p-orbital. Stability increases with charge dispersal via hyperconjugation, resonance, and +I effect. Full order: Tropylium > Benzylic ≈ Allylic > 3° > 2° > 1° > Methyl — and resonance always beats hyperconjugation when both are present.
⚗️ Click the card to explore
A carbocation is a positively charged carbon species with only 6 valence electrons and three covalent bonds — no complete octet. The central carbon is sp² hybridised, giving trigonal planar geometry with 120° bond angles (similar to boron compounds). Perpendicular to the plane sits a completely empty p-orbital — a powerful Lewis acid / electron sink. Nucleophiles attack from either face equally, which is why carbocation intermediates cause racemisation.
Geometry: sp² · Trigonal planar · Bond angles ~120°
Empty p-orbital ⊥ to the plane → Lewis acid site
From least to most stable:
Methyl < 1° < 2° < 3° < Allylic ≈ Benzylic < Tropylium (aromatic)
The driving force in every case is charge dispersal — concentrated positive charge is high energy; “smeared” charge is low energy. Think of it as the “rich neighbour” rule: the more alkyl groups or π-systems surrounding the cation, the more they share the electron burden.
1. Inductive Effect (+I): Alkyl groups are slightly electron-donating through σ-bonds — they push electron density toward the cation. Weak but cumulative. A 3° cation has three donors; methyl has none.
2. Hyperconjugation: More powerful. Adjacent C–H sigma bond orbitals overlap with the empty p-orbital, delocalising electron density. Count alpha-hydrogens: 3° = 9 H → 9 interactions; 2° = 6; 1° = 3; methyl = 0.
3. Resonance: The strongest effect. If a π-system is adjacent, the positive charge is fully delocalised across multiple atoms. In the allyl cation, charge splits as +0.5 on C1 and C3. In benzylic cations, charge spreads throughout the ring. Tropylium (C₇H₇⁺) gains aromatic stabilisation (6π, Hückel’s rule) — the most stable carbocation of all.
When multiple factors are present, always apply this strict order:
Aromaticity > Resonance (+M) > Hyperconjugation > Inductive Effect (+I / −I)
Halogen paradox: Halogens have both –I (destabilising, through σ-bonds) and +M (stabilising, lone pair donation into empty p). Net effect: still stabilising if directly bonded, but weaker than O or N donation because higher electronegativity reduces lone pair availability. F > Cl > Br > I for p-orbital donation (size match with carbon’s 2p orbital).
Electron-withdrawing groups (–I, –M): –NO₂, –CF₃, carbonyl (C=O) all pull electron density away from an already electron-poor carbon — catastrophic.
Increased s-character: Stability order → alkyl > alkenyl > alkynyl. s-Orbitals are held tightly by the nucleus; more s-character = more electronegative = less able to bear positive charge. Vinylic and alkynyl cations are extremely unstable.
Bridgehead positions (Bredt’s Rule): Bridgehead carbocations cannot adopt the required trigonal planar geometry → massive strain → essentially impossible to form. Similarly, cyclopropyl and cyclobutyl cations suffer severe angle strain.
Anti-aromaticity: Cyclopentadienyl cation (4π electrons) is anti-aromatic — delocalisation actually raises energy rather than lowering it.
Carbocations are the rate-determining step of SN1 and E1. By the Hammond Postulate, a more stable carbocation = lower transition state energy = faster reaction. This is why 3° substrates react far faster than 1° in SN1.
Whenever a less stable cation forms adjacent to a more stable position, a 1,2-hydride or 1,2-methyl shift occurs instantly. Example: 3-methyl-2-butanol + HBr → 2° carbocation at C2 → hydride shift → 3° carbocation at C3 → Br⁻ attacks → major product is 2-bromo-3-methylbutane, NOT 3-bromo-3-methylbutane.
Allyl
(aromatic)
| Carbocation Type | α-H (hyperconjugation) | Main stabilisation |
|---|---|---|
| Methyl (CH₃⁺) | 0 | None |
| Primary (1°) | 3 | Weak +I / hyperconjugation |
| Secondary (2°) | 6 | Moderate hyperconjugation |
| Tertiary (3°) | 9 | Strong hyperconjugation |
| Allylic / Benzylic | varies | Resonance (charge delocalised) |
| Tropylium (C₇H₇⁺) | — | Full aromaticity (6π) |
In the high-stakes landscape of organic chemistry, understanding the "motion picture" of a reaction requires a deep dive into its most fleeting characters: reaction intermediates.
Among these, the carbocation is arguably the most critical.
Whether you are navigating an SN1 substitution, an E1 elimination, or an electrophilic addition to an alkene, your ability to predict the major product hinges entirely on your mastery of the carbocation stability order.
A carbocation stability order dictates that reaction intermediates decrease in stability in the sequence: tropylium (aromatic) > Benzyl ≈ allyl > tertiary (3°) > secondary (2°) > primary (1°) > methyl. This thermodynamic hierarchy is governed by electron delocalisation through resonance, hyperconjugation, and positive inductive effects (+I) that disperse the electron-deficient carbon's positive charge.
Carbocation Stability Order:
The stability of carbocations follows the order: Tertiary (3°) > Secondary (2°) > Primary (1°) > Methyl
| Stability Factor | Mechanism | Examples |
| Resonance | Delocalisation of charge | Benzylic, Allylic |
| Hyperconjugation | Sigma-bond orbital overlap | Tertiary, Secondary |
| Inductive Effect | Electron donation via bonds | Alkyl groups |
Key Takeaway: Stability is determined by the ability of adjacent groups to disperse the positive charge. Highly substituted carbocations are more stable due to the combined effect of inductive donation and hyperconjugation.
Table of Contents
What is a carbocation? (The Electron-Deficient Species)
Structure and Hybridisation: Defining the sp² hybridised, trigonal planar carbon.
A carbocation, historically termed a "carbenium ion", is a positively charged species featuring a carbon atom with only six valence electrons and three covalent bonds.
Because it lacks a complete octet, it is a high-energy, transient species that occupies a local minimum on a potential energy reaction coordinate diagram.
Experimental evidence, including X-ray crystallography, confirms that the central carbon of the vast majority of carbocations is sp²-hybridised.
This electronic configuration results in a trigonal planar geometry, in which the three substituents are orientated at the corners of an equilateral triangle, with bond angles of approximately 120°.
This structural arrangement is remarkably similar to neutral boron compounds, which are also trivalent and electron-deficient.
The Empty p-Orbital: Why Carbocations are Powerful Lewis Acids.
Perpendicular to the plane created by the three sigma (σ) bonds lies an unoccupied, non-bonding p-orbital.
This empty orbital acts as a powerful "electron sink" or Lewis acid, making carbocations extremely reactive toward Lewis bases (nucleophiles).
The electrostatic potential diagrams for these species clearly show this cationic centre in blue, highlighting the exact spot where a nucleophile will attack.
Due to this planar geometry, nucleophiles can attack from either face of the empty orbital, which is a crucial consideration for stereochemical outcomes.
The Standard Carbocation Stability Order Hierarchy
To predict the major product in organic mechanisms, mastering the standard order of carbocation stability is indispensable. Thermodynamically, gas-phase and solution-phase experiments confirm that highly substituted alkyl cations always outperform lower-order congeners.
The reactivity of various carbocation types is primarily dictated by how well the positive charge is dispersed.
As a general chemical principle, concentrated charge is unstable (high-energy), whereas dilute or "smeared" charge is stable (low-energy).
When comparing simple alkyl systems, the observed carbocation stability order is: Tertiary (3°) > Secondary (2°) > Primary (1°) > Methyl
The "Rich Neighbour" Analogy: How alkyl groups donate electron density.
As an educator, I often tell my students: "When you are electron-poor, it helps to have rich neighbours."
In a carbocation, adjacent alkyl groups act as these "rich neighbours" by donating electron density to the electron-deficient centre.
Core Electronic Factors Governing the Carbocation Stability Order
Inductive Effect (+I): Short-range electron pushing.
One mechanism for this donation is the inductive effect. Carbon is slightly more electronegative than hydrogen, meaning C–H bonds bear a small dipole where the carbon is partially negative.
In an alkyl group, these small dipoles add up, allowing the carbon of the alkyl group to donate electron density to the adjacent carbocation.
A tertiary cation, with three such alkyl groups, can disperse its charge far more effectively than a methyl cation, which is attached only to hydrogen atoms.
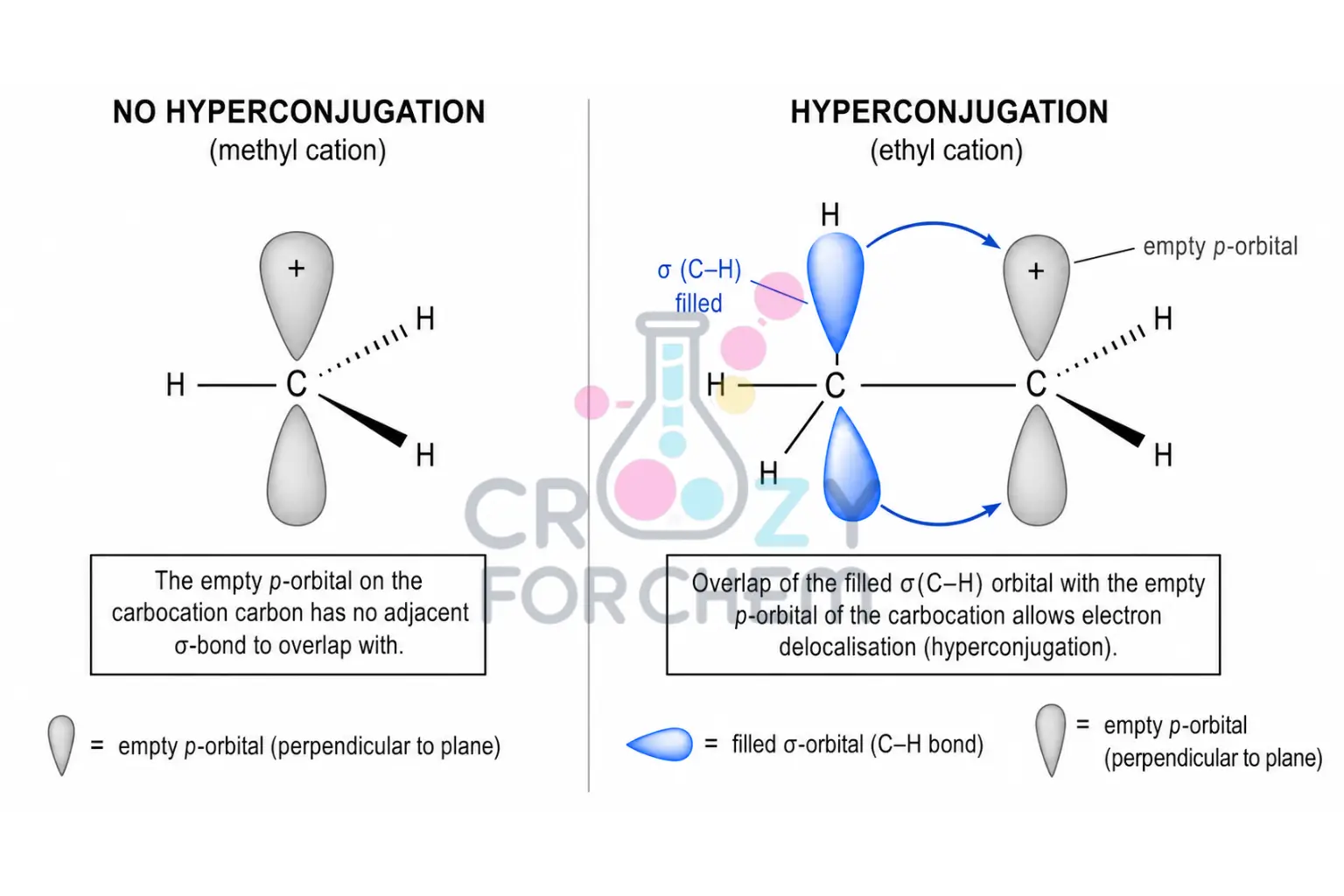
Hyperconjugation stabilisation through σ-bond overlap.
A more sophisticated and powerful explanation is hyperconjugation.
This involves the parallel overlap of the empty p-orbital with adjacent hybridised orbitals participating in sigma bonds (typically C–H or C–C).
Electrons from these neighbouring sigma bonds are shared with the p-orbital, providing significant stabilisation.
- Tertiary cations possess nine α-hydrogens, allowing for nine hyperconjugative interactions.
- Secondary cations have six, and primary cations have three. Strong physical evidence for this model is seen in the adamantyl carbocation, where C–C bonds aligned with the empty p-orbital are noticeably lengthened (weakened) while the bond at the cationic centre is shortened, gaining partial double-bond character.
The Power of Resonance in Stability
While alkyl substitution is a foundation of the carbocation stability order, resonance stabilisation is often the "trump card" that provides even greater energy reduction.
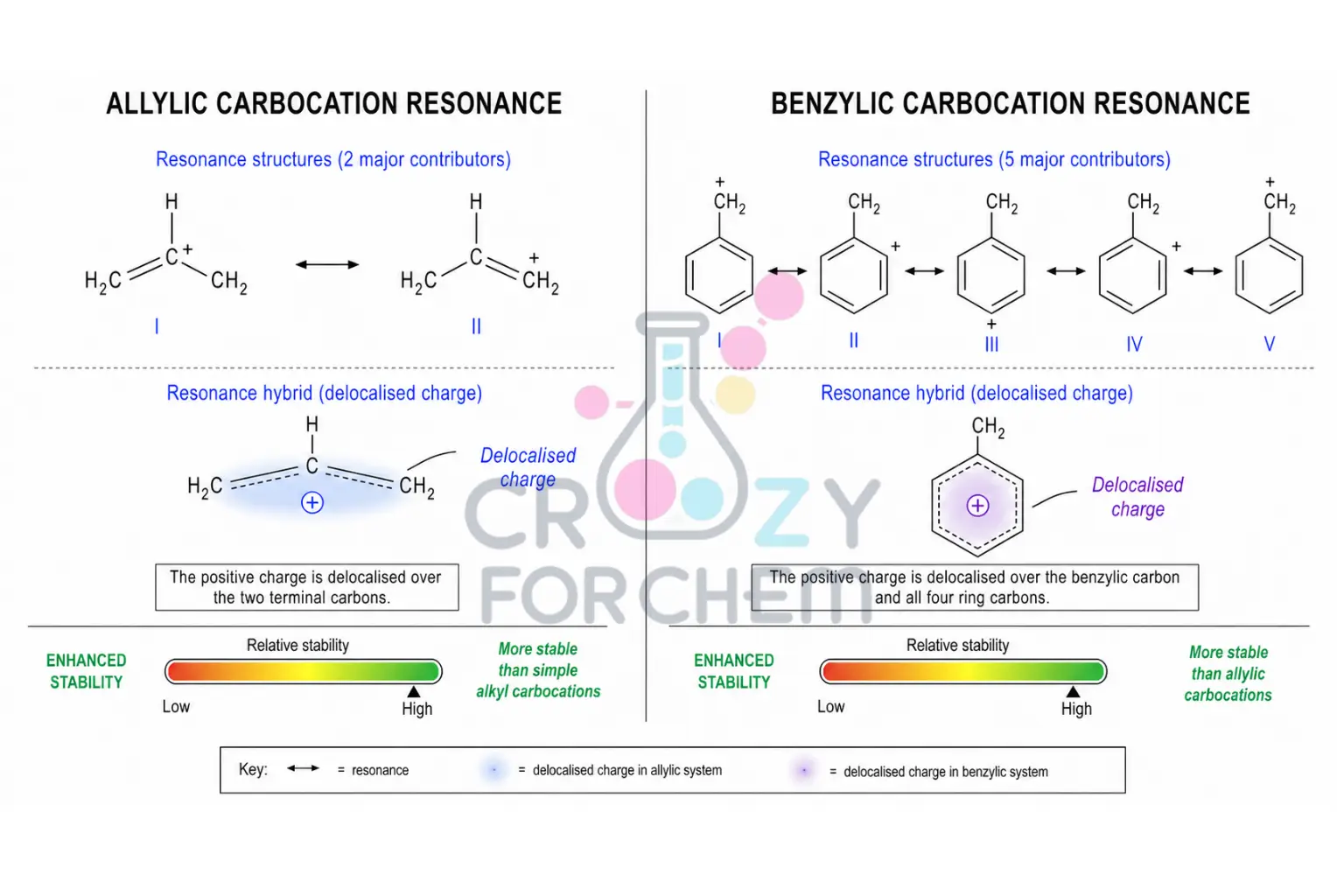
Allylic and Benzylic Carbocations: Delocalisation as the Ultimate Stabiliser.
If a pi (π) system is adjacent to the cationic centre, the positive charge can be "smeared" over multiple atoms through resonance stability.
- An allylic carbocation (where the charge is next to a double bond) allows the charge to be shared between multiple carbons. For example, in the allyl cation, the charge is shared between C-1 and C-3, giving each a charge density of +0.5 instead of +1.
- A benzylic carbocation (adjacent to a benzene ring) is even more stable, as the charge is delocalised throughout the aromatic ring. Experimental evidence shows that a primary benzylic carbocation is roughly as stable as a tertiary t-butyl cation. If the resonance is extensive enough, as in the trityl cation (Ph₃C⁺), the species is so stable that it forms a crystalline salt that can be stored on a shelf indefinitely.
Aromatic Stability: Special cases like tropylium and cyclopropenium.
Certain cyclic carbocations gain extraordinary stability through aromaticity. The tropylium ion (C₇H₇⁺) and the cyclopropenium ion (C₃H₃⁺) satisfy Hückel's Rule (4n+2 π-electrons) and are exceptionally stable.
Conversely, the cyclopentadienyl cation is anti-aromatic and unusually unstable because delocalisation in its 4π system actually increases its electronic energy.
Stabilisation by Adjacent Lone Pairs
It is a common mistake to assume that electronegative atoms like oxygen or nitrogen always destabilise cations. If these atoms are directly adjacent to the positive centre, they can donate a lone pair to form a new π-bond.
The Full Octet Rule
The result of this "pi-donation" is that every atom in the resulting resonance structure, including the carbon, obtains a full octet.
Even though the heteroatom carries a formal positive charge, the stabilisation gained from providing the carbon with a full octet is immense.
- Heteroatom Trends: The ability to donate is proportional to basicity and inversely proportional to electronegativity. Thus, nitrogen is a better pi-donor than oxygen, which is better than fluorine.
- Orbital Overlap: Going down the periodic table, donation ability decreases as orbital size increases. For example, fluorine is a better donor to carbon than iodine because its 2p orbitals match the size of the carbon's 2p orbital perfectly, whereas the larger 5p orbitals of iodine overlap poorly.
The "Hierarchy of Effects": Predicting Success
When multiple factors influence a molecule, you must follow a clear hierarchy to determine the final carbocation stability order:
- Aromaticity: Provides the highest stability.
- Resonance (Mesomeric Effect): Generally more powerful than inductive or hyperconjugative effects.
- Hyperconjugation: Usually stronger than simple inductive effects.
- Inductive Effect: The weakest force, though direct inductive effects can sometimes dominate if other factors are equal.
The Halogen Paradox: Balancing Induction and Mesomeric Effects.
In halogens, the negative inductive effect (-I) often outweighs the positive mesomeric effect (+M) in terms of overall reactivity, making them electron-withdrawing overall, although resonance still dictates the orientation of many reactions.
Factors that Destabilise Carbocations
Just as some groups provide a "cushion" for the charge, others make the cation essentially impossible to form.
Electron-Withdrawing Groups (EWG)
Strongly electronegative groups that cannot donate a lone pair, such as -CF₃, -NO₂, or a nearby carbonyl group (C=O), pull electron density away from the positive centre, greatly increasing its potential energy.
An adjacent carbonyl is particularly destabilising because any resonance form would leave oxygen with only six valence electrons, which is highly unstable.
s-Character, Bridgehead Strain, and Bredt’s Rule.
The stability of carbocations decreases as the s-character of the carbon increases: alkyl (most stable) > alkenyl > alkynyl (least stable).
Because s-orbitals are closer to the nucleus, increasing s-character makes the carbon nucleus more electronegative and less able to bear a positive charge. This makes vinylic carbocations very unstable.
Bridgehead Positions and Small Rings
Bridgehead carbocations are notoriously unstable because they cannot attain the ideal trigonal planar geometry. This violation of Bredt's Rule creates massive strain.
Similarly, carbocations on small rings like cyclopropanes and cyclobutanes are unstable due to angle strain.
Measuring Carbocation Stability
Hydride affinity and ionisation rates as quantitative benchmarks.
To put these carbocation stability trends on a quantitative basis, chemists use specific measurements:
- Hydride Affinity: The energy released when a carbocation reacts with a hydride ion (H⁻) to form a neutral molecule. A lower hydride affinity indicates a more stable cation.
- Ionisation Rates: Measuring the rate of hydrolysis of alkyl halides in polar protic solvents, where faster rates correlate with more stable intermediates.
Carbocation Rearrangement: The Drive Toward Stability
Why 1,2-hydride and methyl shifts define the major product.
Because carbocations are high-energy species, they are prone to carbocation rearrangement via 1,2-hydride shifts or 1,2-alkyl shifts. This occurs provided the shift generates a more stable carbocation.
For example, a secondary carbocation will rapidly rearrange into a tertiary one, often resulting in products with a completely different carbon skeleton than the "predicted" starting structure.
Worked Example: 1,2-Hydride Shift
Q: What is the major product when 3-methyl-2-butanol is treated with HBr?
Step 1: HBr protonates the OH group → water leaves → secondary carbocation forms at C-2.
Step 2: The adjacent C-3 bears a methyl group and a hydrogen. A 1,2-hydride shift occurs — the hydrogen migrates from C-3 to C-2, generating a more stable tertiary carbocation at C-3.
Step 3: Br⁻ attacks the tertiary carbocation.
Major Product: 2-bromo-3-methylbutane (not the "expected" 3-bromo-3-methylbutane)
💡 Exam Tip: Whenever you see a secondary carbocation adjacent to a tertiary carbon, always check for a 1,2-hydride or methyl shift – this is the most common trap in JEE Organic Chemistry.
Carbocation Stability Order: JEE Main & NEET Exam Guide
For students preparing for JEE Main, JEE Advanced, and NEET, carbocation stability is one of the highest-yield topics in organic chemistry, appearing almost every year in multiple forms.
The Non-Negotiable Stability Order (Memorise This)
Methyl < Primary (1°) < Secondary (2°) < Tertiary (3°) < Allylic ≈ Benzylic < Aromatic (Tropylium)
Priority Rule for Conflicting Effects (JEE Advanced)
When two effects oppose each other, follow this strict hierarchy:
- Aromaticity (highest)
- Resonance / Mesomeric Effect (+M)
- Hyperconjugation
- Inductive Effect (+I or −I)
💡 JEE Trick: If a carbocation has both resonance AND inductive effects acting on it, resonance always wins. A primary benzylic carbocation is more stable than a tertiary alkyl carbocation for exactly this reason.
3 Most Frequently Asked JEE/NEET Question Types:
Type 1 — Direct Stability Ranking Q: Arrange the following in order of increasing stability: methyl carbocation, t-butyl carbocation, allyl carbocation, benzyl carbocation. A: Methyl < t-Butyl < Allyl ≈ Benzyl
Type 2 — Hyperconjugation Counting Q: How many hyperconjugative structures does the t-butyl carbocation have? A: Nine – three methyl groups × three C–H bonds each = 9 alpha-hydrogens = 9 hyperconjugative interactions.
Type 3 — Halogen Paradox (JEE Advanced) Q: Is CH₂⁺–F more or less stable than a primary alkyl carbocation? A: More stable, despite fluorine's strong −I effect, its lone pair donation (+M) via p-orbital overlap provides net stabilisation. However, this stabilisation is weaker than oxygen or nitrogen donation because of fluorine's high electronegativity, reducing lone pair availability.
Conclusion: Why Carbocation Stability Order Dictates Reaction Speed
Mastering the carbocation stability order is the key to understanding chemical kinetics. The formation of the carbocation is typically the rate-determining step of a reaction.
According to the Hammond Postulate, a more stable intermediate leads to a lower-energy transition state, resulting in a lower activation energy and a faster reaction.
By recognising the structural "safety nets" resonance, substitution, and aromaticity, you can predict with precision how a molecule will transform.
As an educator, I tell my students: don't just memorise the trends; look for the "rich neighbours" and delocalisation pathways that make an electron-deficient carbon atom bearable.
Frequently Asked Questions
-
Q1: What is the correct carbocation stability order?
The generally accepted carbocation stability order is: Tertiary (3°) > Secondary (2°) > Primary (1°) > Methyl. This trend is determined by the ability of adjacent groups to disperse the positive charge through induction, hyperconjugation, and resonance.
-
Q2: Why is a tertiary carbocation more stable than a primary one?
A tertiary (3°) carbocation is more stable because it has three alkyl groups attached to the central carbon. These groups provide inductive electron donation and, more importantly, hyperconjugation, where electrons from adjacent C–H sigma bonds overlap with the empty p-orbital, effectively "diluting" the positive charge.
-
Q3: What makes allylic and benzylic carbocations so stable?
Allylic and benzylic carbocations benefit from resonance stabilisation. In these species, the positive charge is not fixed on a single atom but is delocalised (or "smeared") over the adjacent π system. This delocalisation significantly lowers the potential energy of the intermediate compared to a simple alkyl carbocation.
-
Q4: Can electronegative atoms like nitrogen or oxygen stabilise a carbocation?
Yes, counterintuitively, they can! If a nitrogen or oxygen atom is directly adjacent to the cationic centre, its lone pair can be donated to form a new π-bond. This gives every atom in the resulting resonance structure a full octet, which is a powerful stabilising force that often outweighs the electronegativity of the atom.
-
Q5: What are the main factors that destabilise a carbocation?
A carbocation is destabilised by:
Electron-Withdrawing Groups (EWG): Groups like -NO₂ or -CF₃ pull electron density away from an already electron-poor centre.
Increased s-Character: Vinylic and alkynyl carbocations are unstable because s-orbitals are held tightly by the nucleus.
Geometric Strain: Bridgehead carbocations (Bredt's Rule) are highly unstable because they cannot adopt the required trigonal-planar geometry. -
What is the carbocation stability order in the context of JEE and NEET examinations?
For competitive examination purposes, the complete carbocation stability order, from least to most stable, is: Methyl < Primary (1°) < Secondary (2°) < Tertiary (3°) < Allylic ≈ Benzylic < Tropylium (aromatic). In JEE Advanced questions, this order is often complicated by the simultaneous presence of resonance and inductive effects. In such cases, the hierarchy of effects aromaticity > resonance > hyperconjugation > inductive effect must be applied strictly to arrive at the correct answer.
-
Why does carbocation rearrangement occur, and how does it affect the major product?
Carbocation rearrangement occurs because carbocations are high-energy, unstable intermediates that will always seek the lowest possible energy state. When a 1,2-hydride shift or 1,2-methyl shift can convert a less stable carbocation into a more stable one, this migration happens spontaneously and extremely rapidly. The consequence for reaction products is significant; the major product may have a completely different carbon skeleton from what a straightforward mechanism would predict. In examinations, this is one of the most frequently tested concepts in SN1 and E1 reaction mechanisms.