
A carbocation rearrangement is a 1,2-shift where a less stable intermediate transforms into a more stable one. Hydride shifts are kinetically preferred over methyl shifts, following migratory aptitude: H > Ph > 3° > 2° > 1° alkyl. Because carbocations are sp² trigonal planar, nucleophilic attack from either face leads to racemisation — the original chiral configuration is completely lost.
⚗️ Click the card to explore
Carbocations have an incomplete octet — only 6 valence electrons — making them highly unstable. A 1,2-shift occurs only when the resulting cation is instantly more stable than the original. Think of it as a relay race: the migrating group is the baton, and the molecule passes it the moment doing so gives an energy advantage.
Stability order: 3° > 2° > 1° > methyl carbocation
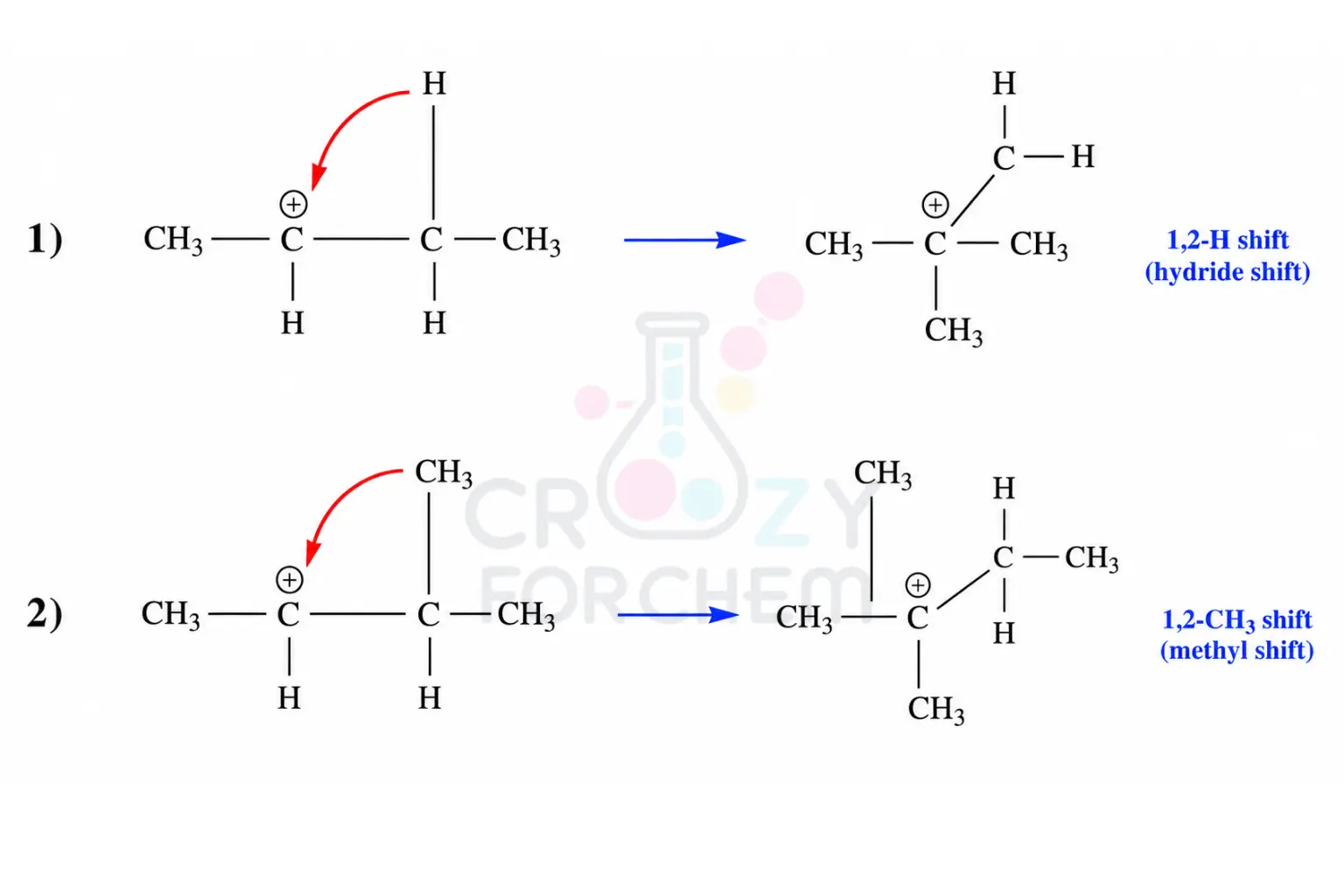
A 1,2-hydride shift moves H⁻ (hydrogen with its bonding pair) to the adjacent carbocation. It is almost always preferred because hydrogen has the highest migratory aptitude and moves fastest. A 1,2-methyl shift moves an entire –CH₃ group and only occurs when the neighbouring carbon is quaternary and has no available hydrogen.
Migratory aptitude: H > Ph > 3° alkyl > 2° alkyl > 1° alkyl (CH₃ lowest)
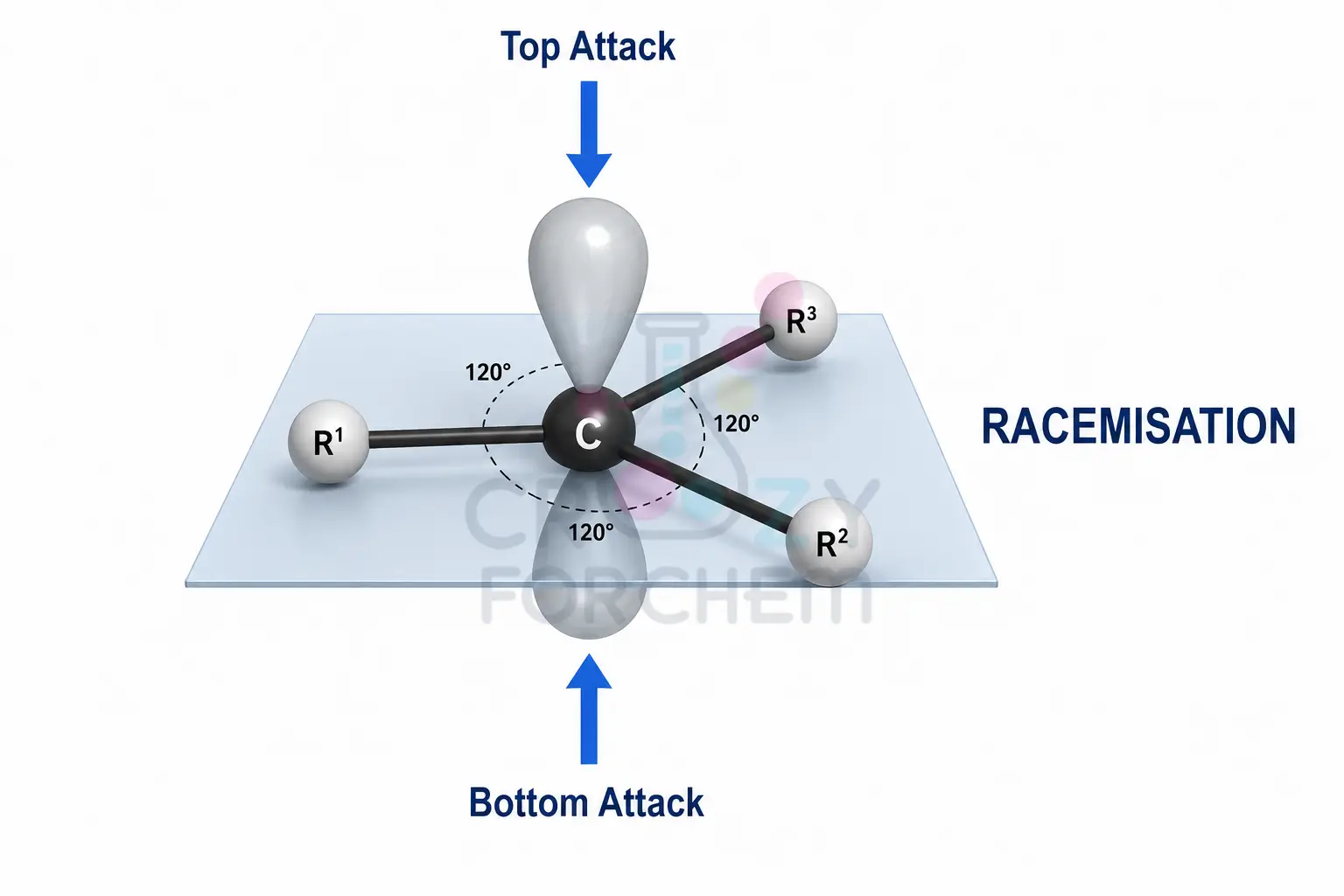
When a carbocation forms, carbon rehybridises from sp³ to sp², becoming trigonal planar and flat. A vacant p-orbital sits perpendicular to this plane. The nucleophile has equal probability of attacking from the top or bottom face — giving a 50/50 mixture of enantiomers (racemic mixture). Any pre-existing chirality is completely erased.
Adjacent only: Shifts are strictly 1,2 — only to the immediately neighbouring carbon. No 1,3 or longer shifts (poor orbital overlap).
Resonance trumps substitution: A 2° cation will shift to a benzylic or allylic position even if substitution level stays the same — delocalisation is a powerful driving force (Wagner–Meerwein).
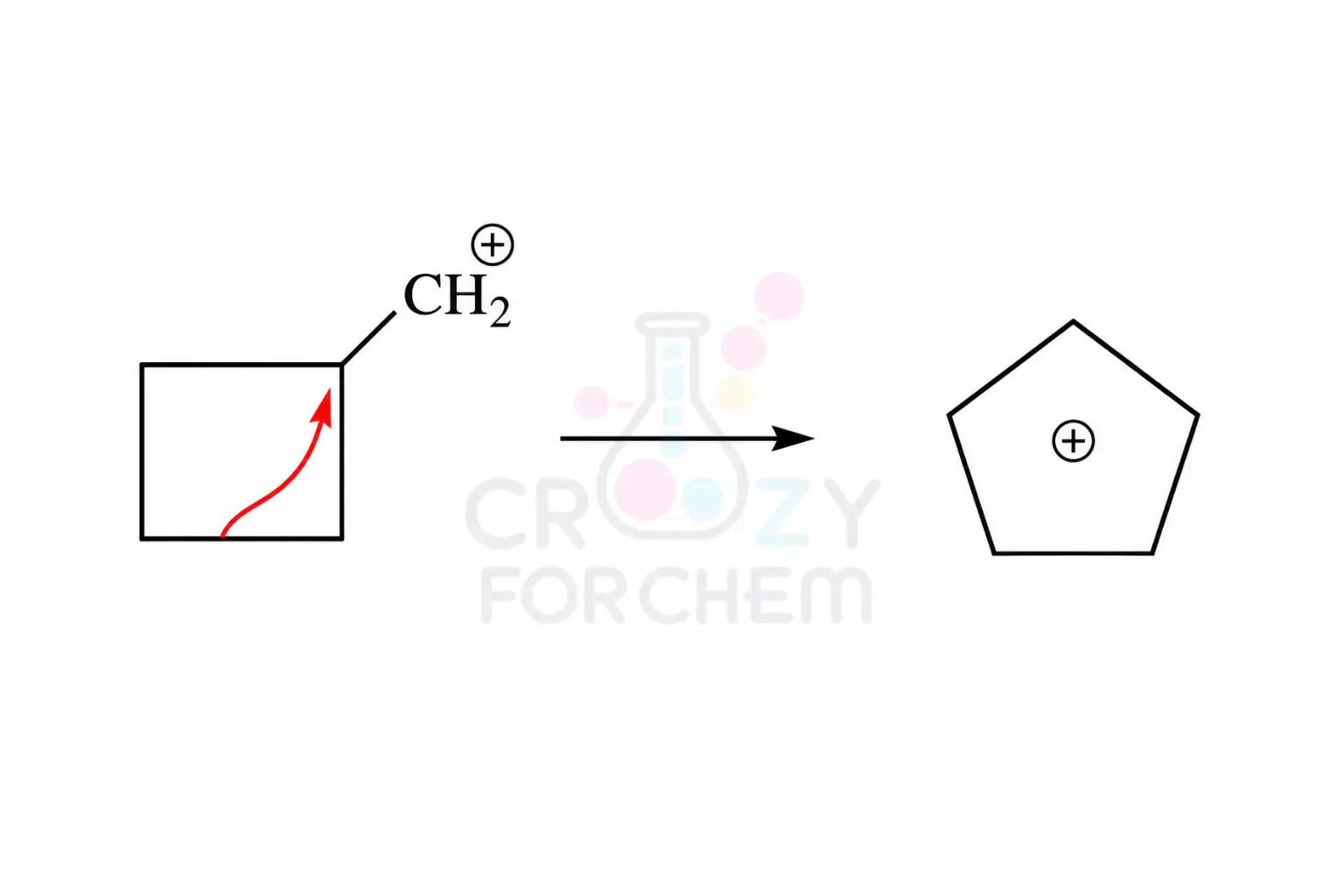
Ring expansion: Cyclobutane rings expand to cyclopentane to relieve angle strain via a carbocation shift.
Rearrangements only occur in SN1 and E1 reactions, which go through a free carbocation intermediate. In SN2 and E2, the mechanism is concerted — no carbocation forms, so no rearrangement is possible. Approximately 90% of molecules undergoing SN1 rearrange before the nucleophile attacks.
shifts allowed
hybridisation
rearrange first
→ racemic mixture
trigonal planar
driving rearrangement
| Group | Migratory Aptitude | Reason |
|---|---|---|
| Hydride (H⁻) | Highest | Small size, fastest kinetics |
| Phenyl (Ph) | High | Electron-rich π system |
| 3° Alkyl | Medium | Bulky but stable |
| Methyl (CH₃) | Lowest | Slowest migration |
In the high-stakes landscape of organic chemistry, the ability to predict the outcome of a reaction is what separates an amateur from an expert. One of the most fascinating, yet often frustrating, phenomena for students is the stereochemistry and rearrangement of carbocations.
As an educator, I tell my students that hybridisation is like mixing colours to get a new shade; in the same way, carbon atoms can reorganise their bonding and spatial arrangement to find the most stable configuration possible.
When a molecule forms a carbocationic intermediate, it does not always stay in its original configuration; instead, it often undergoes a molecular “shuffle” to find a lower-energy state. Mastering this process is the master key to accurate major product prediction in complex organic mechanisms.
Table of Contents
The Driving Force: Why do Carbocations Rearrange?
The Quest for Stability
Carbocations are inherently unstable, high-energy species because they possess an incomplete octet, having only six valence electrons.
They act as local minima on a potential energy diagram, seeking ways to increase their stability through electronic effects.
A rearrangement, specifically a 1,2-shift mechanism, will occur if, and only if, the resulting carbocation is instantly more stable than the original one.
Typically, this means a shift from a less substituted to a more substituted centre, such as a secondary (2°) carbocation rearranging into a tertiary (3°) or a resonance-stabilised benzylic cation.
Tertiary carbocations, being the most stable among alkyl groups, generally do not feel the “urge” to rearrange further unless a resonance-stabilised position is nearby.
The “Relay Race” Analogy
To visualise this, think of the 1,2-shift mechanism as a relay race. The transition state represents that split second where both carbons have a “hand” on the migrating group (the baton).
As the bonding pair of electrons interacts with the empty p-orbital of the carbocation, the bond to the original carbon weakens while the new bond starts to form. Once the “baton” is passed, the positive charge is transferred to the adjacent carbon, creating a new, more stable species.
Methyl Shift vs Hydride Shift: Deciphering the Mechanism
When a carbocation seeks a more stable state, it primarily utilises two “tools”: the 1,2-hydride shift and the 1,2-methyl shift. Deciding which occurs depends on the migratory aptitude of the groups attached to the vicinal (neighbouring) carbon.
1,2-Hydride Shifts
In a 1,2-hydride shift, a hydrogen atom migrates along with its bonding pair of electrons to the adjacent positive centre.
This is often the preferred pathway because hydrogen has a high migratory aptitude and migrates faster than alkyl groups. This shift is common when a secondary carbocation is adjacent to a tertiary carbon bearing at least one hydrogen.
1,2-Methyl (Alkyl) Shifts
When no suitable hydrogen atoms are available, such as when the adjacent carbon is quaternary (bonded to four other carbons), the molecule undergoes a 1,2-methyl shift. Here, an entire methyl group (sometimes called a methide ion) jumps to the next carbon.
This methyl shift and hydride shift in the carbocation stability context explains why a secondary carbocation might transform into a tertiary one by moving a methyl group from a neighbouring quaternary centre.
The Hierarchy of Migration
A critical rule for major product prediction is understanding the hierarchy of migratory aptitude: H > Phenyl > 3° Alkyl > 2° Alkyl > 1° Alkyl. When comparing a methyl shift vs a hydride shift in carbocation stability, the hydride shift is almost always preferred because it is kinetically faster.
However, if a quaternary carbon is the only neighbour, the methyl group is forced to move to relieve the electronic “stress” of the secondary cation.
| Group | Migratory Aptitude | Reason |
| Hydride (H⁻) | Highest | Small size, faster kinetics. |
| Phenyl (Ph⁻) | High | Electron-rich π system. |
| 3° Alkyl | Medium | Bulky but stable. |
| Methyl (CH₃⁻) | Lowest | Slowest migration. |
Stereochemistry and Rearrangement of Carbocations
Their unique geometry dictates the stereochemistry of carbocations. Understanding the stereochemistry and rearrangement of carbocations requires looking at the electronic configuration of the central carbon.
Trigonal Planar Geometry
When a carbocation forms, the carbon transitions from sp³ to sp² hybridisation. This results in a trigonal planar geometry where the molecule is flat, with substituents separated by 120°. Crucially, there is a vacant, non-bonding p-orbital that sits perpendicular to this plane.
Educator’s Insight: “Students often ask me if π-bonds affect hybridisation. Remember, π-bonds are formed by unhybridised p-orbitals, so they are never counted in hybridisation. Only count σ-bonds and lone pairs!”.
Racemisation and Loss of Optical Activity
Because the intermediate is flat, a nucleophile has a 50/50 chance of attacking from the “front” or the “back” of the p-orbital. This leads to racemisation, the formation of a nearly equal mixture of enantiomers.
Consequently, any original chiral configuration is “erased” the moment the planar carbocation forms. As I tell my students, the “dash and wedge” notations of the starting material should be discarded in your intermediate drawings because the 3D tetrahedral structure has been lost.
Expert Rules for Predicting Rearrangements
The “Adjacent Only” Rule
One of the most important rules in the stereochemistry and rearrangement of carbocations is that shifts are strictly limited to vicinal (neighbouring) carbons.
While students often wonder if a 1,3 or 1,4 shift can occur to reach a distant stable spot, this is generally prohibited because orbital overlap must occur between the C-H bond and the empty p-orbital.
Carbocations are “blind” to long-term gains; they only move if the immediate next step provides a boost in stability.
Resonance as a “Trump Card”
Resonance stabilisation is a powerful driving force for the Wagner-Meerwein rearrangement.
A secondary carbocation will readily rearrange to a benzylic or allylic position, even if the substitution level does not technically increase, because the delocalisation of charge across a π-system offers significant energetic gains.
A Wagner-Meerwein rearrangement is a 1,2-shift of a hydrogen or alkyl group to an adjacent carbocation, resulting in a more stable intermediate. It is the fundamental mechanism behind 1,2-hydride and methyl shifts.
Ring Expansion
In cyclic systems, rearrangements often take the form of ring expansion. This occurs to relieve ring strain, particularly in four-membered rings like cyclobutanes, which can expand into more stable five-membered cyclopentane rings before the nucleophile attacks.
Avoiding Common Synthesis Pitfalls
The 90% Rule
One of the most vital takeaways for any SN1 mechanism is that rearrangements are sneaky and fast.
It is estimated that rearrangements occur in about 90% of molecules before any nucleophile has a chance to attack. If you forget to check for a potential shift, you will likely predict the wrong product.
SN1 vs SN2 Outcomes
Carbocation rearrangements are a hallmark of unimolecular reactions such as the SN1 mechanism and E1 elimination. In contrast, concerted SN2 and E2 reactions happen “all at once”, meaning no free carbocation is formed, and thus no rearrangement can occur.
Classic research by Frank C. Whitmore on neopentyl systems showed that when these molecules are forced into a carbocation pathway, they invariably rearrange to find stability.
Conclusion: Mastering the Molecular Shuffle
Mastering the stereochemistry and rearrangement of carbocations is essential for understanding the logical flow of organic chemistry.
By recognising the trigonal planar geometry of sp² intermediates and applying the rules of migratory aptitude, you can predict racemisation and accurately identify the major product.
As you navigate these mechanisms, always remember to “number your carbons” and check the vicinal positions.
If a secondary cation sees a “rich neighbour” (a tertiary or quaternary carbon) next door, it will almost certainly leap to find a more stable home. Once you account for these sneaky shifts, the complex puzzle of organic synthesis begins to fall into place.
Frequently Asked Questions (FAQs)
-
Why does a 1,2-hydride shift occur in carbocations?
A 1,2-hydride shift occurs to increase the stability of a carbocation intermediate. This rearrangement involves the migration of a hydrogen atom, along with its bonding electron pair, from an adjacent carbon to the positive centre. It typically happens when a less stable secondary (2°) carbocation can be converted into a more stable tertiary (3°) or resonance-stabilised cation.
-
Between a methyl shift and a hydride shift, which is preferred?
Generally, a hydride shift is preferred over a methyl shift due to its higher migratory aptitude. Hydrogen is smaller and kinetically faster to migrate. A methyl (alkyl) shift usually only occurs when the adjacent carbon is quaternary (bonded to four carbons) and has no available hydrogen atoms to facilitate a hydride shift.
-
What is the driving force behind carbocation rearrangement?
The primary driving force is the attainment of a lower energy state through increased stability. Carbocations rearrange to move the positive charge to a more substituted carbon atom, where it can be better stabilised by inductive effects, hyperconjugation, or resonance (delocalisation).
-
How does carbocation rearrangement affect stereochemistry?
Carbocation rearrangement leads to racemisation. Since the intermediate carbocation has a trigonal planar geometry with an empty p-orbital, the nucleophile can attack from either side with equal probability. This results in the loss of original optical activity and the formation of a racemic mixture of enantiomers.
-
Can a carbocation rearrange more than once?
Yes, multiple rearrangements can occur as long as each subsequent step results in a more stable intermediate. However, “blind” shifts that result in a less stable cation or maintain the same stability without a resonance gain are energetically unfavourable and generally do not occur.
-
Does rearrangement occur in SN2 reactions?
No, carbocation rearrangements do not occur in SN2 mechanisms. SN2 is a concerted, one-step process where the nucleophile attacks as the leaving group departs. Since no free carbocation intermediate is formed, there is no opportunity for a 1,2-shift to take place.
-
What is a carbocation rearrangement?
A carbocation rearrangement is a process where a molecular structure reorganises itself to transition from an unstable state to a more stable one. This typically occurs through a 1,2-shift mechanism, where an adjacent atom or group migrates with its bonding electrons to the electron-deficient carbon centre.
-
What is the difference between a methyl shift and a hydride shift?
1,2-Hydride Shift: A hydrogen atom moves with its bonding pair of electrons to the adjacent positive carbon.
1,2-Methyl Shift: An entire methyl group (methide ion) migrates to the neighbouring positive carbon when no suitable hydrogens are available on a quaternary centre. Generally, hydride shifts occur more frequently because hydrogen has a higher migratory aptitude and moves faster than alkyl groups. -
What is the order of migratory aptitude in carbocation rearrangement?
The general tendency for groups to migrate follows a specific hierarchy: Hydrogen > Phenyl group > Tertiary alkyl > Secondary alkyl > Primary alkyl. In most practical settings, if a molecule has a choice between a hydride and a methyl shift to achieve the same stability, the hydride shift will occur first.
-
Why do carbocations only undergo 1,2 shifts?
Rearrangements are strictly limited to adjacent carbons because orbital overlap must occur between the migrating bond and the empty p-orbital of the carbocation. While a 1,3 or 1,4 shift might theoretically lead to a more stable spot, the molecule cannot “see” past the immediate next carbon; it only moves if the very next step provides an instant boost in stability.
-
How does carbocation rearrangement affect stereochemistry?
Because carbocations are sp²-hybridised and trigonal planar, they are flat structures. This geometry means a nucleophile can attack from either the front or the back with equal probability, leading to racemisation (a 50/50 mixture of enantiomers). Any original chiral configuration is effectively lost the moment the carbocation forms.
-
Do rearrangements occur in SN2 reactions?
No, carbocation rearrangements are exclusive to unimolecular reactions (SN1 and E1). In a concerted SN2 mechanism, the nucleophile attacks at the same time the leaving group departs, so no free carbocation is ever formed for a rearrangement to take place.
-
What is the driving force for a Wagner-Meerwein rearrangement?
The driving force is always the attainment of a more stable carbocation. This usually involves moving from a primary or secondary carbocation to a more substituted tertiary one, or to a position stabilised by resonance (such as benzylic or allylic positions).
-
How common are carbocation rearrangements?
In reactions where a carbocation intermediate is formed, it is estimated that 90% of molecules will undergo rearrangement before reacting with a nucleophile. This makes identifying potential shifts critical for accurate major product prediction.